
Introducing the 'Hallmarks of Aging' Series
A dozen reasons why we get older.


Why do we get older?
This is a question that’s been pondered by philosophers for centuries and aging biologists for decades — and a question that will likely continue to invigorate research for untold years to come.
While we haven’t identified a single root cause of aging, science has allowed us to untangle several processes that are consistently associated with it — these have been coined the “Hallmarks of Aging.”
The field of aging research has seen significant progress since the publication of the original "Hallmarks of Aging" in 2013, with nearly 300,000 articles exploring the topic. A decade later, the need for an updated edition of the hallmarks has emerged to incorporate the latest insights into the mechanisms underlying the aging process. We know much more than we did before (or do we?)1
Aging “hallmarks” are defined by three primary criteria: the time-dependent manifestation of aging-related alterations, the ability to accelerate aging through experimental manipulation, and the potential to slow down or reverse aging through therapeutic interventions.
In other words, to be a hallmark, something must change with time, reliably cause aging, and slow down aging in its absence/manipulation.
In 2013, 9 hallmarks were identified: DNA instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient-sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication.
Over the past decade, these hallmarks have gained further validation through research on various organisms, including mice and non-human primates, confirming their relevance to mammals (that’s us!)
Three new hallmarks have been identified and added to the list: disabled macroautophagy, chronic inflammation, and dysbiosis.
The integration of these new hallmarks into the original 9 provides a comprehensive framework for understanding the complex mechanisms underlying aging and age-related diseases.
In this ongoing series, we’ll discuss each of the dozen hallmarks and identify novel, up-and-coming interventions that might impact them to slow the aging process.
A brief warning — some of this info will be very technical. However, I hope that you follow along with interest as we dive into cutting-edge biology and explore why we age, and what we can do about it.

Today, we tackle the first two hallmarks: genomic instability and telomere attrition.
Genomic instability: an overview
Genomic instability refers to the susceptibility of an organism's genetic material, or genome, to undergo alterations due to a myriad of external and internal factors.
These factors include agents such as chemicals, physical forces, and biological agents, as well as endogenous challenges like errors in DNA replication, issues with chromosome separation, oxidative stress, and spontaneous chemical reactions. The consequences of these challenges are genetic abnormalities — point mutations, deletions, translocations, telomere shortening, single- and double-strand breaks, rearrangements of chromosomes, disruptions in nuclear architecture, and even gene mutations caused by the integration of viruses.
These alterations contribute to the development of certain disruptive patterns in an organism's genome, which can play a role in both normal aging and age-related diseases.
To counteract genetic instability, we’ve evolved intricate systems of DNA repair and maintenance. These mechanisms work to mitigate the damage inflicted on nuclear DNA and mitochondrial DNA (mtDNA) and to uphold the structural integrity and stability of chromosomes. However, with time, the efficiency of these DNA repair networks diminishes, leading to the accumulation of genetic damage. With age, we’re worse at repairing the ever-accumulating damage.
Accumulating DNA damage not only contributes to the aging process but also results in the accumulation of DNA fragments in the cytosol of cells. This phenomenon underscores the intricate relationship between genomic instability and the broader aging process, shedding light on the importance of DNA repair mechanisms in maintaining the integrity of our genome.

Genomic instability affects nuclear DNA, mitochondrial DNA, and nuclear architecture.
Nuclear DNA within cells experiences the accumulation of somatic mutations and other forms of damage as we age. Such DNA damage can lead to the malfunctioning of cells, ultimately disrupting tissue and overall organismal equilibrium.
Of particular significance is the impact on stem cells, where DNA damage can hinder their role in tissue rejuvenation or cause their depletion, driving aging and increasing our vulnerability to age-related disorders.
Remarkably, normal human tissues amass a substantial number of mutations throughout life, with young individuals' cells already harboring hundreds of mutations, potentially exceeding 2,000 mutations per cell by middle age.
Across species, there appears to be an inverse correlation between somatic mutation rates and lifespan. While the normal rate of mutation fixation isn't definitively linked to aging, evidence underscores the role of DNA repair deficiencies in promoting aging.
DNA repair mechanism alterations in mice accelerate aging and underlie certain human progeroid (pro-aging) syndromes. On the other hand, certain transgenic mice with heightened expression of DNA repair enzymes have extended healthy lifespans. Similar observations emerge from studies involving humans and other long-lived species, where improved DNA repair mechanisms coevolve with increased longevity. For this reason, interventions targeting nuclear DNA's mutational load or repair mechanisms might delay aging and age-related ailments, but more research is needed.

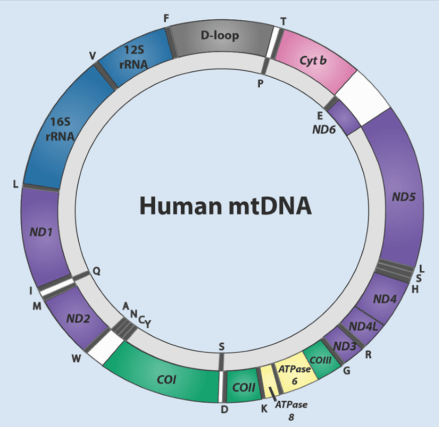
Mitochondrial DNA (mtDNA) is the genetic material found within the mitochondria, the energy-producing organelles of cells. mtDNA plays a vital role in cellular functions and metabolism, but it’s highly prone to mutations due to factors such as a high rate of replication, limited repair mechanisms, exposure to oxidative stress, and lack of protective histones.
As we age, mutations and deletions in mtDNA accumulate, potentially contributing to the aging process and age-related diseases. While the impact of these mutations on functional aging remains uncertain, studies have shown that these mutations often arise from replication errors rather than oxidative stress (another cause of aging that we’ll explore later on).
Studies in mice have linked mtDNA damage to accelerated aging and reduced lifespan, particularly in relation to deletions in mtDNA. This suggests that addressing mtDNA mutations could potentially extend healthspan and lifespan.
Nuclear architecture refers to the structure and organization of the cell nucleus, where our genetic material resides. The nuclear lamina is a framework that anchors chromatin (DNA and proteins) and other complexes within the nucleus. Defects in the nuclear lamina can lead to genome instability as well.
Accelerated aging conditions like Hutchinson-Gilford and Néstor-Guillermo progeria syndromes are associated with mutations in genes that encode components of the nuclear lamina. Levels of lamin B1, another protein associated with the nuclear lamina, decrease during cellular senescence. Studies on animal and cellular models have unveiled the response mechanisms triggered by nuclear lamina abnormalities, including the activation of the tumor suppressor protein p53 and changes in adult stem cells.

Various interventions, including systemic injection of substances and gene editing strategies, have been explored to address nuclear lamina abnormalities. While these approaches hold promise for progeria treatment, their impact on normal aging is not yet supported by evidence.
Fixing genomic instability
As far as this aging hallmark goes, interventions to reduce its effects are sparse and have included overexpression of certain genes (i.e., BubR1 or SIRT6) or the application of certain anti-cancer treatments.

Mutations to our genetic material seem inevitable, and as such, genomic instability is considered one of the primary hallmarks of aging.
Telomere attrition: an overview
Telomere attrition refers to the gradual shortening of the protective DNA sequences called telomeres located at the ends of chromosomes — these are sometimes analogized to the protective caps on the ends of our shoelaces.
